Data Collection
ASPREE was supported by a sophisticated data system known as the ASPREE Web Accessible Relational Database (AWARD) suite. The AWARD suite consists of four communicating modules: AWARD-Data, AWARD-General, AWARD-Adjudicator and AWARD-Access Management System (AMS) (1). Each module consists of a Web Client Application linked to a secure SQL database. The functions of each module are shown in Figure 1 below. All data collection for ASPREE was facilitated by one of the four modules of the AWARD suite and included collection of operational fields (e.g. visit due date and booked date) and analytical fields (e.g. height, date of myocardial infarction (MI)).
Analytical data was annotated for quality via AWARD using a commentary code system. For more information on commentary codes please see Quality Control within the section About the Data Set. Further information on AWARD and data quality in the ASPREE Clinical Trial has been published elsewhere (1).
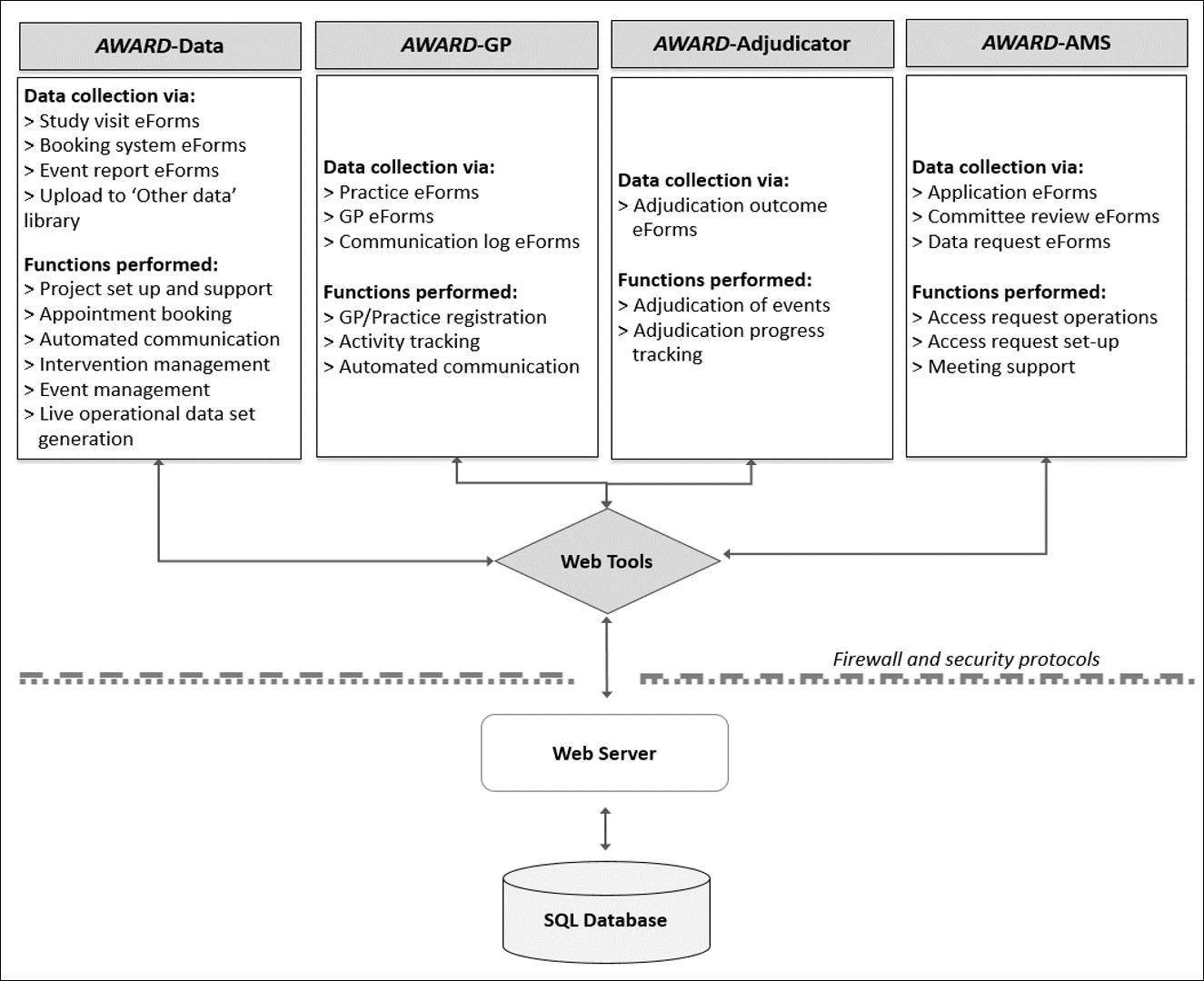
The AWARD suite SQL database is hosted in the Monash University Data Centre in Clayton and within the Monash ‘Red Zone’. This is a specialised facility with tightly controlled data access and storage. All data is encrypted at 2048 bits in transit via SSL through the web server and IP Sec tunnels to the database cluster. The data centre is secured and monitored electronically and can only be entered by authorised personnel. Data centre processes are detailed by Monash University’s Information Security Management System (ISMS) which is Certified to ISO 27001 international standards (Certification No: ITGOV40017). These are continually reviewed and improved as a part of the continuous improvement process of the ISMS. The Data House at Clayton (Victoria) is mirrored to a second centre at Noble Park (Victoria) to provide redundancy. Only system administrators have unrestricted access to the ASPREE database, no users have direct access.
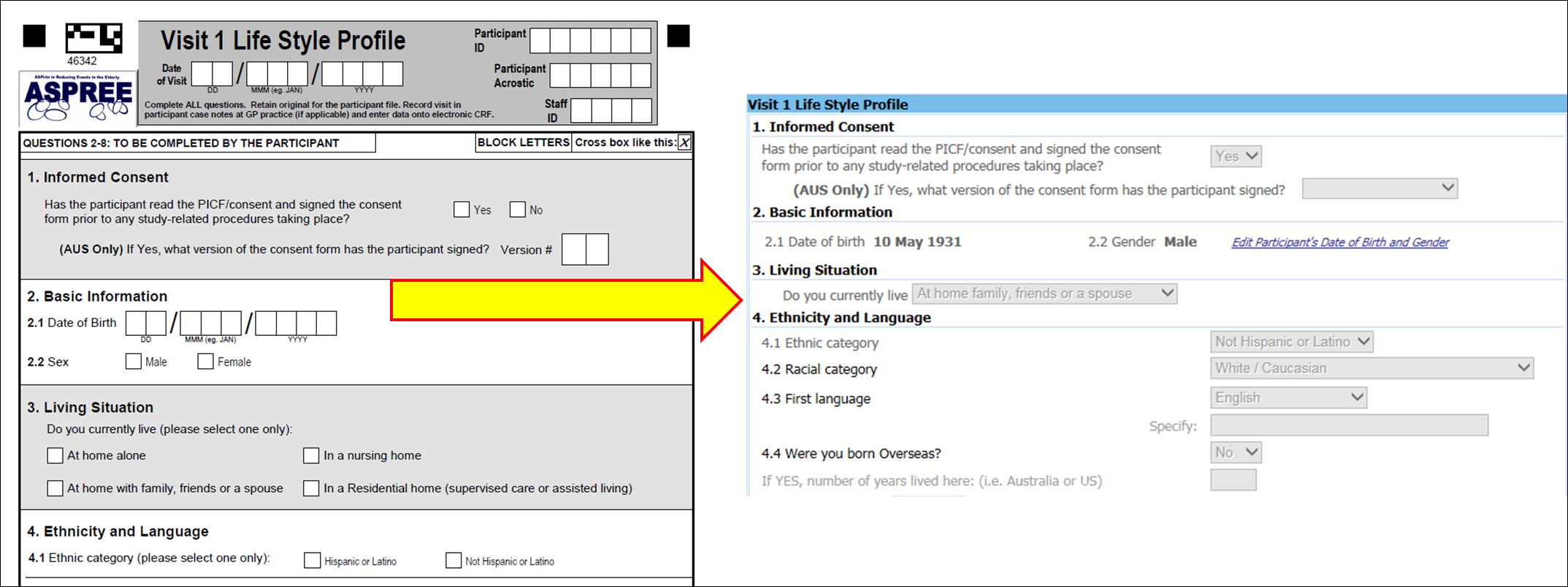
Operational and analytical data collected on case report forms (CRFs) were entered into AWARD via the web application by study staff. Each visit conduced in the field was created in the web application as an electronic visit. Following creation of the electronic visit, staff entered the data from each CRF into the electronic case report form (eCRF) on AWARD-Data (see Figure 2 below).
Data Collection during ASPREE, the Bridge and ASPREE-XT
During the ASPREE Clinical Trial, cognitive function was measured at baseline, annual visits 1, 3 and 5, and Milestone visits (which could have occurred as any one of annual visit 3 through 7 depending on the participants’ duration in the trial). A 30 minute cognitive assessment battery was used and included the Modified Mini-Mental State Examination (3MS) (2), Symbol-Digit Modalities Test (SDMT) (3), Hopkins Verbal Learning Test—Revised (HVLT-R) (4), and the Controlled Oral Word Association Test (COWAT) (5). During ASPREE-XT, the 3MS was conducted annually while the COWAT, SDMT, HVLT-R and Color Trails were collected every year from the second ASPREE-XT annual visit (XT02) onward.
During the ASPREE Clinical Trial and ASPREE-XT, depression was measured each year using the CES-D 10 assessment tool (a self-reported questionnaire) (6).
During the ASPREE Clinical Trial, physical function was measured at baseline, annual visits 2, 4, 6 and Milestone visits using the performance-based measures of gait speed and hand grip tests. Self-reported ADL and instrumental activities contained within the LIFE Disability Questionnaire (7), and quality of life was measured using the Short Form 12 (SF-12) (8) were collected annually . During ASPREE-XT, all physical function measures were conducted annually.
Laboratory measures included hemoglobin and urine albumin creatinine ratio (ACR). If a urine ACR value was high (based on a cut-off of 2.5mg/mmol for males or 3.5mg/mmol for females), the primary care provider (PCP)/General Practitioner (GP) was notified and asked to request a repeat test in order to confirm microalbuminuria. Completion of this additional test was at the discretion of the PCP/GP.
Hospitalisation for reasons other than primary or secondary endpoints were also captured during both the ASPREE Clinical Trial and ASPREE-XT.
During the ASPREE Clinical Trial, study medication was recorded as described below. After the cessation of study medication in June 2017, questions relating to current aspirin use were introduced to the remaining Milestone visits, six-month phone calls and annual visits.
Data Collection Schedule
The ASPREE Clinical Trial Measurement and Study Activity schedule is summarised in Figure 3 and the ASPREE-XT schedule is summarised in Figure 4.

X indicates when each category of measures was carried out. Superscripts a, b, or c specify the tests within each category that were performed at the designated time point. Lack of superscript indicates that all category measures were carried out at the designated time point. ** Milestone visit took place in years 3 to 7 depending on year of randomisation.

X indicates when each category of measures was carried out. Superscript β indicates where a test or consent form may have been mailed to the participant ahead of the study visit or γ administer by phone; * alcohol and smoking information; # height was measured at XT03 visit only (if not collected at XT03 visit, height collection attempted at next in-person visit), $ waist circumference was measured from XT02 visit onward.
Questionnaire Design
Validated questionnaires were utilised where possible. These included:
- Short Form 12 (SF-12) (8);
- Modified Mini-Mental State Examination (3MS) score ≤77 (2);
- Hopkins Verbal Learning Test-Revised (HVLT-R) (4);
- Controlled Oral Word Association Test (COWAT) (5);
- Symbol-Digit Modalities Test (SDMT) (3);
- Center for Epidemiologic Studies-Depression (CES-D) assessment tool, Version 10 (6); and
- LIFE Disability Questionnaire (7).
ASPREE developed CRFs to capture demographics, lifestyle information, physical examination results, concomitant medication (ConMed) use, clinical events, and study medication compliance.
In-Field Data Collection
During the ASPREE Clinical Trial, Bridge period and ASPREE-XT, in-field data collection was conducted by trained staff during in-person assessments with participants. Data collection was driven by national standard operating procedures (SOPs) for visit conduct and individual test administration. To ensure that SOPs were understood and implemented correctly, new staff underwent an intensive three to five-day training course when they first commenced at ASPREE. Prior to this training week, staff were asked to view the online training videos and read all relevant visit conduct and test administration SOPs.
During training, staff were informed of the principles underpinning Good Clinical Practice (GCP), the ethical considerations of undertaking research involving human subjects, and guidance on how to collect informed consent. All visit-related questionnaires and data collection forms, and in particular the cognitive assessments, were reviewed in detail and rigorously practiced to allow for immediate feedback. During the training schedule, staff were assessed by the ASPREE nominated neurologist on cognitive test administration, and required a pass to independently conduct the cognitive tests. Staff then observed the conduct of ASPREE visits, for a period of three to four weeks, including conducting small portions of the visit themselves, under the guidance of an experienced staff member.
ASPREE trainers formally accredited successful staff members for independent visit conduct by assessing their competency to accurately collect all relevant data, and successfully enter it into the AWARD-Data system, in compliance with visit conduct SOPs and GCP.
Conduct of In-Field Data Collection
In-field data collection occurred at participant visits with ASPREE staff. These visits were booked at the participant’s local GP practice, a community venue, a clinical trial study site or at their home. To conduct a study visit, the following equipment and documentation was required:
- A printout of the Annual Visit Summary Report (AVSR) (re-named as the Annual Visit Summary Checklist (AVSC) during ASPREE-XT)
- Relevant visit-specific source documentation (i.e. CRFs) pack
- Annual study medication from the ASPREE study medication cabinet (during the ASPREE Clinical Trial only)
- A working blood pressure (BP) monitor (and spare batteries) and both medium and large BP cuffs
- Timer (and spare batteries)
- Calculator
- Ruler or equivalent
- Pathology provider request forms (if required)
- Weighing scales
- Figure Finder tape measure for abdominal circumference
- Stop watch (to 1/100 sec) for 3m gait speed test
- Retractable tape measure for 3m gait speed test
- Masking tape (24 mm wide) for 3m gait speed test
- Hand grip dynamometer
The AVSR/AVSC was a summary of relevant information previously entered into AWARD-Data. The report was available for download via AWARD-Data and provided participant-specific information without the need to bring primary participant files to the study visit. Staff recorded relevant information about family medical history, clinical events, ConMed use and study medication use on the AVSR/AVSC and then transcribed the information into AWARD-Data at the time of data entry.
Potential Errors During In-Field Data Collection
While equipment was serviced regularly, occasionally a device malfunction prevented collection of data at a given visit. Where device error was the reason for missing data, a commentary code of 4 was applied (see Quality Control within the section About the Data Set).
If an annual visit was missed for any reason (e.g. the participant was away for an extended period of time) data collection linked with the missed visit was conducted at the next annual visit. For example, if the missed visit was a Year 2 annual visit, grip strength and gait speed tests would be administered at the Year 3 visit. In this situation, the date of the Year 2 annual visit is the same as the Year 3 annual visit date. Data that is collected at each annual visit and hence cannot be ‘caught up’ is left blank for the missed annual visits (i.e. Year 2 annual visit in this example). Where data is missing due to the conduct of a visit at the same time as another visit a commentary code of 12 was applied.
If any data collection field was missed due to staff error (e.g. not taking the correct CRFs to a visit, failing to administer a mandatory measure etc.), a commentary code of 5 was applied.
Collection of Clinical Events
Scheduled collection of ASPREE clinical events occurred at all six-month phone calls and annual visits through completion of the following forms and assessments:
- Six-Month Phone Call form (ASPREE only)
- Six-Month Life Disability form
- Annual Visit Life Disability Questionnaire
- Annual Visit 3MS (and CES-D 10)
- Annual Visit ConMeds form
- Annual Visit Participant Medical History Update form (PMHU) [note that during ASPREE-XT this changed to being a six-month PMHU]
- Annual Visit review of medical records
Participants were able to contact ASPREE at any time and report a clinical event, which was recorded in AWARD-Data, for follow-up either prior to or at the time of the next annual visit. During ASPREE-XT, the PMHU form was completed at each six-month call.
Death could be detected at any point in time. In Australia, linkage with the Ryerson Index of obituaries was performed on a weekly basis to detect deaths. A search of the NDI was conducted in both countries in November 2017. Two additional linkages were conducted in Australia in 2019, 2020 and 2022.
Clinical Event Collection
At 6M phone calls during the ASPREE Clinical Trial and the Bridge period, participants answered a series of seven questions regarding ASPREE endpoints (i.e. cancer, clinically significant bleeding, dementia, depression, hospitalisation for heart failure, MI, stroke), one question regarding other hospitalisations, 19 structured questions about new diagnoses of specific medical conditions of interest, and one open question about other new diagnoses. This part of the six-month phone call mirrored the questions on the PMHU administered at annual visits conducted during the same timeframe. As such, clinical event reporting was consistent at all scheduled collection time points during the ASPREE Clinical Trial and Bridge period.
During ASPREE-XT, questions regarding new diagnoses of specific medical conditions of interest were reduced to 13 structured questions. Data relating to these questions were captured from April 2019 onward. An additional question relating to completion of an Aged Care Assessment (Australia) was added to both the six-month phone call and annual visit PMHU in June 2019. The annual visit PMHU form was updated to capture other hospitalisations from April 2019, and six-month phone calls were updated to include other hospitalisation admissions from August 2019. Hospitalisation data was captured via proxy where the six-month phone call was conducted with a suitable proxy. Hospitalisation data for the first 14 months of ASPREE-XT were captured retrospectively.
Clinical events reported via the standard questions at six-month phone calls or PHMU at annual visits were subject to collection of supporting documentation and adjudication. Although not an ASPREE primary or secondary endpoint, cause of death was also subject to collection of supporting documentation and adjudication.
Fact of death was subject to confirmation with two independent sources (e.g. family and General Practitioner/Primary care provider (GP/PCP), and published obituary/funeral notice).
Data Driven Event Collection
Participants could trigger for data driven events at six-month phone calls and annual visits. Data driven events included:
- Persistent physical disability (detected via the LIFE Disability Questionnaire)
- Depression (detected via the CES-D 10 questionnaire)
- Dementia (detected via the 3MS assessment, entry of a dementia-specific medication or completion of a dementia-specific clinical assessment)
For persistent physical disability endpoint, participants were asked a list of six modified Katz ADL questions related to difficulty performing daily activities at six-month phone calls, annual visits and Milestone visits. A response of ‘a lot of difficulty’ or ‘unable to perform’ an activity, or the need for assistance to complete the activity was considered a trigger for the physical disability endpoint. If an equivalent response (i.e. ‘a lot of difficulty’ or ‘unable to perform’ an activity, or need for assistance) was reported for the same activity at re-administration approximately six months later, the trigger was considered persistent and the physical disability endpoint was confirmed.
For the depression endpoint, participants were asked to complete the CES-D 10 questionnaire at Year 1, 3, and 5 visits between 1 March 2010 and 31 December 2014, and at all annual visits from 1 January 2015 onwards. Any assessment with a score of eight or more was considered a depression endpoint. The determination of depression endpoint sub-type (i.e. incident, recurrent or persistent) was automated based on previously entered CES-D 10 data. If all previous CES-D 10 scores were < 8, the event was considered to be incident. If one or more previous CES-D 10 scores were 8+ but the CES-D 10 score immediately preceding was < 8, the event was considered recurrent. If the CES-D 10 score immediately preceded was also 8+, the event was considered persistent.
For the dementia endpoint, endpoint triggers were driven by the completion of the 3MS assessment at an ASPREE study visit, detection of dementia-specific medications by study staff, or the identification of dementia-specific clinical assessments that were conducted outside of the ASPREE study. The 3MS assessment was completed at baseline and re-administered at the Year 1, 3 and 5 visits and the Milestone visit. During ASPREE-XT, the 3MS was completed at each annual visit. A predicted long-term 3MS score was calculated for each participant based upon their raw score at baseline, age and education. A post-randomisation 3MS score that was below 78 (out of 100) or was greater than 10.15 drop from the participant’s predicted long term 3MS score, generated a dementia endpoint trigger based on the CES-D 10 result (see Table 1 below). The presence of depression can hinder cognitive performance, therefore if depressive symptoms were detected via the CESD-10, alongside declined cognitive performance, a reassessment was conducted three months later to allow time for the depressive symptoms to resolve.
In response to the practical limitations experienced during the global COVID-19 pandemic, in which in-person visits were temporarily ceased, study workflows were adapted to enable collection of cognitive data at annual visits conducted via phone call. This included modified 3MS, COWAT and HVLT-R assessments. This change was introduced in April 2020 in Australia, and in July 2020 in the US, for all participants who usually attended annual visits in person. The change was also introduced for participants in both countries who usually completed annual visits via phone call in November 2020. When conducted via phone call, the overall maximum possible score for the 3MS was reduced from 100 to 74, as questions 7, 11, 12, 13 and 14 could not be conducted due to the requirement of physical input from the participant. As a result, the 3MS trigger for further dementia assessment was reduced to a score below 54, and participants could no longer trigger due to a 10.15 drop from their predicted 3MS score when the 3MS was administered by phone. Any 3MS reassessments for phone-call triggers were conducted via phone call (again with a reduced overall maximum possible score of 74 and a score below 54 as a trigger).
Table 1. Dementia endpoint trigger decision rubric based on CES-D 10 score at baseline and time of 3MS examination conduct.
| Baseline CES-D 10 result | 3MS completed in person | 3MS completed via phone call | Dementia endpoint trigger? |
|---|---|---|---|
| CES-D 10 result linked with 3MS <78 or >10.15 drop from predicted score | CES-D 10 result linked with 3MS < 54 | ||
| <8 | <8 | Yes | |
| 8+ | <8 | Yes | |
| <8 | 8+ | No* | |
| 8+ | 8+ | Yes | |
*participant required re-administration of 3MS and CES-D 10 in three months’ time.
To accommodate those with unresolved depression not present at baseline, a dementia endpoint was triggered if a participant’s baseline CES-D 10 was less than eight and three consecutive CES-D 10 scores of more than eight were subsequently obtained.
Dementia triggers were followed up by the Dementia Assessment team. Where possible, genuine dementia triggers resulted in an in-person dementia assessment visit. This visit included administration of the Alzheimer Disease Assessment Scale-Cognitive subscale (ADAS-Cog) (9) (10) (for aphasia, apraxia), Color Trails (11) (for executive functioning; not conducted if completed at the most recent annual visit), Confusion Assessment Method proforma short form (12) (13) (for delirium), visual agnosia (for agnosia) (14), and Alzheimer’s Disease Cooperative Study–Instrumental Activities of Daily Living scale (ADCS IADL) (15) based on both self-report and surrogate information (for functional decline). This set of cognitive tests were purposefully curated to ensure major cognitive domains were tested. The Dementia Endpoint Adjudication Committee utilised these test results but the data have not been included in the data set.
Event coding and adjudication is outlined in the section Endpoints and Adjudication.
Clinical event trigger data has been included in Sections B5, C1, E1 and E2 of the ASPREE Longitudinal Data Set (Version 3) and the ASPREE-XT Longitudinal Data Set (XT04).
Study Medication Tracking
ASPREE Clinical Trial study medication was tracked via an online log called the Drug Log in AWARD-Data. At randomisation, each participant was assigned a unique medication number. All subsequent bottles of study medication provided were labelled with this unique number.
New bottles of study medication were dispensed and old bottles retrieved at each annual visit during the ASPREE Clinical Trial. When bottles were retrieved, the following data was collected and entered into the Drug Log for the bottle in question:
- Returned date
- Count of returned pills (set at a value of one if a bottle was not returned)
- If not retrieved, the reason the bottle could not be returned
If a bottle was lost or accidentally destroyed, participants were provided with a new bottle of medication from the emergency medication supply. Data from the Drug Log has been included in Section G1 of the ASPREE-XT Longitudinal Data Set (XT04) and the ASPREE Longitudinal Data Set (Version 3).
Concomitant Medication Collection
ConMeds were collected directly from participants wherever possible. At baseline and each annual visit, participants were asked to bring along all their medications. Prescription medications were recorded on the AVSR/AVSC and entered into AWARD-Data by staff either by selecting an option from a dropdown list of medication or, if the option was not available, entering the medication name in free-text. The start and stop year (if the participant stopped using the medication) was recorded for each medication. At the start of ASPREE-XT (February 2018), data collection of the ConMeds was changed to capture whether the participant was using the medication specifically at the time of the annual visit.
At the conclusion of the ASPREE Clinical Trial, a neural network was trained to ingest free-text ConMed data and produce a list of probable Anatomical Therapeutic Chemical (ATC) medication codes based on publicly available large medication data sets. The list of probable ATC medication codes was then reviewed by two staff who independently recorded the correct ATC code. Discordant cases were reviewed by the Data Manager and resolved (16). Additional free-text ConMed data collected during the Bridge period and up until the second ASPREE-XT annual visit has been included in the ASPREE-XT Longitudinal Data Set (XT04) and were reviewed in a similar manner.
ConMed data, including medication name, ATC code and whether or not the medication was taken during each calendar year of follow-up (during the ASPREE Clinical Trial and Bridge period) or whether or not the participant was taking the medication at the time of the current annual visit (during ASPREE-XT) can be found in Section B5 of the ASPREE-XT Longitudinal Data Set (XT04). A statistical methods guide for the analysis of ConMed data has been prepared to support analysts using the ASPREE ConMed data (17). The use of this document is strongly encouraged.
Pathology Measure Collection
During the ASPREE Clinical Trial, Bridge and ASPREE-XT, the following pathology measures were collected guided by the schedules shown above:
- Hemoglobin (ASPREE Clinical Trial, Bridge and ASPREE-XT) with a Full Blood Examination (FBE; Australia, ASPREE-XT only) or Complete Blood Count (CBC; US, ASPREE-XT only)
- Fasting lipids (total cholesterol, HDL, LDL, triglyceride; ASPREE Clinical Trial and Bridge only)
- Fasting glucose (ASPREE Clinical Trial and Bridge only)
- Serum creatinine
- Urine albumin: creatinine ratio (ACR)
- HbA1c (ASPREE-XT only)
During ASPREE-XT, fasting lipids were not collected and an FBE or CBC was requested from the XT02 visit and onward. In addition, non-fasting glucose was only collected at XT01.
In general, ASPREE did not conduct pathology measures directly (point-of-care hemoglobin measures were available at some US sites). Rather, in Australia, participants were provided with pathology slips and asked to attend a local pathology centre for blood and urine collection and analysis. Study contact details were included in the pathology slip to enable feedback of results to ASPREE. In the US, blood and urine were collected and analysed at the clinic where the study visit was conducted.
If urine ACR was > 2.5 mg/mmol (males) or > 3.5 mg/mmol (females) a letter was sent to the participant’s GP/PCP to recommend follow up with the participant and arrange for a second urine ACR test. The results of these follow up urine ACR tests are in the variables labelled ‘fwACR’. Completion of this additional test was at the discretion of the PCP/GP so this data may be missing.
Results returned to ASPREE were entered into AWARD-Data. Where pathology results were requested from a third-party pathology but never provided, a commentary code of 6 has been applied.
Pathology measure data has been included in Section B4 of the ASPREE-XT Longitudinal Data Set.
References
-
Lockery JE, Collyer TA, Reid CM, Ernst ME, Gilbertson D, Hay N, et al. Overcoming challenges to data quality in the ASPREE clinical trial. Trials. 2019 Dec;20(1):686. doi: 10.1186/s13063-019-3789-2.
-
Teng EL, Chui HC. The Modified Mini-Mental State (3MS) examination. J Clin Psychiatry. 1987 Aug;48(8):314–8.
-
Smith A, Services (Firm) WP. Symbol digit modalities test : manual [Internet]. Los Angeles, Calif. : Western Psychological Corporation; 2002 [cited 2018 Nov 1]. Available from: https://trove.nla.gov.au/version/42656069
-
Benedict RHB, Schretlen D, Groninger L, Brandt J. Hopkins Verbal Learning Test – Revised: Normative Data and Analysis of Inter-Form and Test-Retest Reliability. Clin Neuropsychol. 1998 Feb 1;12(1):43–55.
-
Ross TP. The reliability of cluster and switch scores for the Controlled Oral Word Association Test. Arch Clin Neuropsychol. 2003 Mar;18(2):153–64.
-
Radloff LS. The CES-D Scale: A Self-Report Depression Scale for Research in the General Population. Appl Psychol Meas. 1977 Jun 1;1(3):385–401.
-
LIFE Study Investigators, Pahor M, Blair SN, Espeland M, Fielding R, Gill TM, et al. Effects of a physical activity intervention on measures of physical performance: Results of the lifestyle interventions and independence for Elders Pilot (LIFE-P) study. J Gerontol A Biol Sci Med Sci. 2006 Nov;61(11):1157–65.
-
Ware J, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care. 1996 Mar;34(3):220–33.
-
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984 Nov;141(11):1356–64.
-
Mohs R. Alzheimer Disease Assessment Scale-Cognitive subscale; Adapted from the Administration and Scoring Manual for the Alzheimer’s Disease Assessment Scale, 1994 Revised Edition. The Mount Sinai School of Medicine; 1994.
-
D’Elia L, Satz P, Uchiyama C, White T. Color Trails Test: Professional Manual. Psychological Assessment Resources, Inc; 1996.
-
Inouye SK, van Dyck CH, Alessi CA, Balkin S, Siegal AP, Horwitz RI. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990 Dec 15;113(12):941–8.
-
The Confusion Assessment Method (CAM). Training Manual and Coding Guide. Yale University School of Medicine; 2003.
-
Lezak M. Neuropsychological assessment, 3rd ed. New York: Oxford University Press; 1995.
-
Galasko D, Bennett DA, Sano M, Marson D, Kaye J, Edland SD, et al. ADCS Prevention Instrument Project: assessment of instrumental activities of daily living for community-dwelling elderly individuals in dementia prevention clinical trials. Alzheimer Dis Assoc Disord. 2006 Dec;20(4 Suppl 3):S152-169.
-
Lockery JE, Rigby J, Collyer TA, Stewart AC, Woods RL, McNeil JJ, et al. Optimising medication data collection in a large-scale clinical trial. PLoS One. 2019 Dec 27;14(12):e0226868–e0226868.
-
Broder J, Wolfe J. ConMeds statistical method guidelines v1.0. Melbourne: School of Public Health and Preventive Medicine, Monash University; 2021.